AlphaFold: The Structure Prediction Problem
This is Lecture 7 (first of two parts) of the Protein & Artificial Intelligence course (Spring 2026), co-taught by Prof. Sungsoo Ahn and Prof. Homin Kim at KAIST Graduate School of AI. It covers the structure prediction problem, its significance, and how people use structure prediction today. The companion Lecture 8 covers AlphaFold2's architecture in detail.
Introduction
In November 2020, DeepMind’s AlphaFold2 achieved what many structural biologists had considered a decades-away milestone: predicting protein structures from amino acid sequences at near-experimental accuracy. The result was not a lucky accident. AlphaFold2 encodes deep biological insight—about protein evolution, three-dimensional geometry, and physical symmetry—directly into its neural network architecture.
This lecture asks two questions: why is protein structure prediction important, and how do people use it today? We trace the fifty-year arc from Anfinsen’s thermodynamic hypothesis through CASP14, then examine the practical ecosystem—AlphaFold Database, ColabFold, ESMFold, AlphaFold3—that has democratized structural biology.
Roadmap
| Section | Topic | Why It Is Needed |
|---|---|---|
| 1 | Historical context | Frames the 50-year challenge and the CASP benchmark |
| 2 | Bird’s-eye view | Establishes AlphaFold2’s overall strategy before architectural details |
| 3 | Why structure prediction matters | Connects structure to function, disease, and engineering |
| 4 | How people use structure prediction today | Practical tools, workflows, and limitations |
| 5 | Design principles | Distills the recurring architectural ideas that made AlphaFold2 work |
1. Historical Context: The Fifty-Year Challenge
1.1 Anfinsen’s Thermodynamic Hypothesis
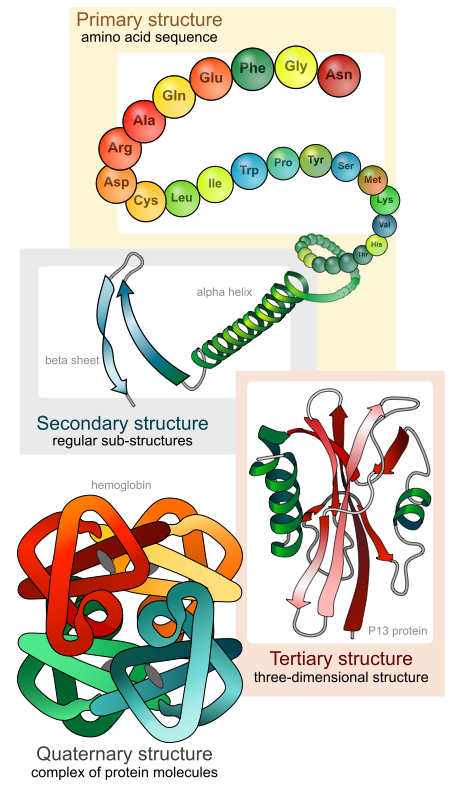
In 1972, Christian Anfinsen received the Nobel Prize in Chemistry for demonstrating that a protein’s amino acid sequence contains all the information necessary to determine its three-dimensional structure1. His experiments on ribonuclease A showed that a denatured2 (unfolded) protein could spontaneously refold into its functional form once the denaturing agent was removed.
The implication was clear: the sequence dictates the fold.
1.2 Levinthal’s Paradox
If sequence determines structure, why can we not simply compute the structure? Cyrus Levinthal pointed out a devastating combinatorial obstacle. A protein of just 100 amino acids, a small protein by any measure, can adopt an astronomical number of backbone conformations. If the protein sampled one trillion conformations per second, it would still take longer than the age of the universe to enumerate them all. Yet real proteins fold in milliseconds.
This is Levinthal’s paradox: the folding process must follow a guided search, not a random one. The energy landscape is “funneled,” steering the chain toward the native state through progressively lower-energy intermediates3. But knowing that a shortcut exists is different from knowing what it is.
1.3 CASP and the Structure Prediction Community
The Critical Assessment of protein Structure Prediction (CASP) competition, launched in 1994, gave the field a rigorous benchmark. Every two years, organizers release protein sequences whose structures have been determined experimentally but not yet published. Prediction groups submit blind predictions, and the results are evaluated against the hidden ground truth.
For 25 years, progress was incremental. Methods improved from roughly 20 GDT-TS (a score where 100 means perfect) in early CASPs to the mid-60s by 2018. Then AlphaFold2 appeared at CASP14 in 2020 and scored a median GDT-TS above 90, crossing the threshold of experimental accuracy for most targets4. A problem that had resisted half a century of effort appeared to yield almost overnight to deep learning.
2. Bird’s-Eye View: How AlphaFold2 Thinks About Proteins
2.1 The Core Insight: No Protein Is Alone
When predicting a protein’s structure, one might think the only available information is the sequence itself—a string of amino acid letters. But every protein has evolutionary relatives: sequences that diverged from a common ancestor and have been independently shaped by natural selection.
These relatives are collected into a multiple sequence alignment5 (MSA), where homologous (introduced in Lecture 5) sequences are arranged so that evolutionarily equivalent positions line up in columns.
Examining an MSA reveals two kinds of signal:
- Conservation. Some positions rarely change because mutations there would break the protein.
- Co-variation. Some positions change together: when position 15 mutates, position 47 compensates. These correlated mutations indicate that the two positions are in physical contact in the folded structure.
AlphaFold2 makes evolutionary information the central organizing principle of its architecture, not merely an input feature.
2.2 AlphaFold2 Pipeline Overview
The following diagram shows the overall architecture of AlphaFold2, from input sequences to 3D structure output.
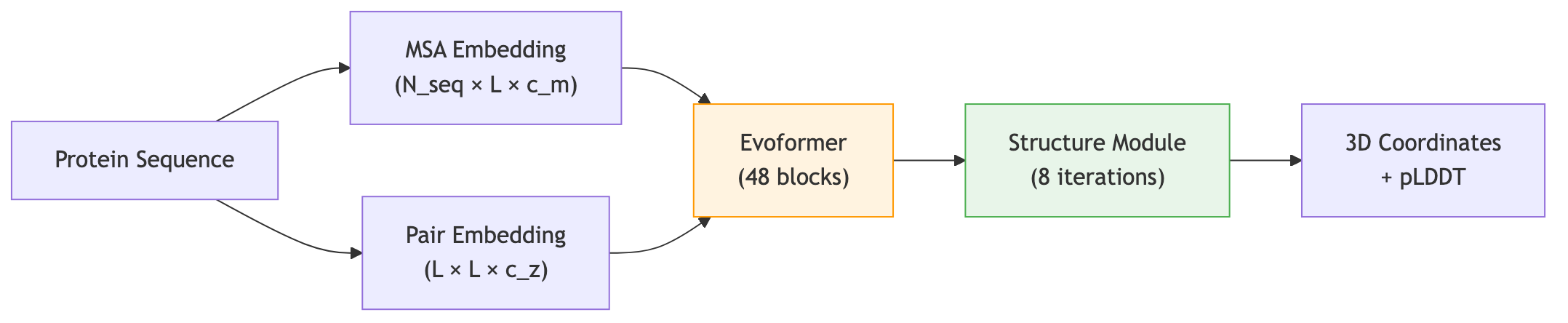
2.3 Two Representations, One Structure
AlphaFold2 maintains two parallel data structures throughout most of its computation:
- The MSA representation has shape \([N_{\text{seq}} \times L \times c_m]\), where \(N_{\text{seq}}\) is the number of aligned sequences, \(L\) is the protein length, and \(c_m\) is the feature dimension. Each entry encodes information about one position in one sequence.
- The pair representation has shape \([L \times L \times c_z]\). Entry \((i, j)\) encodes what the network believes about the relationship between residue \(i\) and residue \(j\)—their spatial proximity, hydrogen-bonding potential, or co-evolutionary coupling.
These two representations communicate throughout the network. Evolutionary signals from the MSA inform pairwise relationships, and pairwise constraints help interpret the MSA. By the end of this exchange, the pair representation contains a detailed predicted distance map.
2.4 From Distances to Coordinates
A distance map tells you which residues are close, but it does not directly specify where they are in three-dimensional space. The Structure Module takes the refined representations and converts them into atomic coordinates. This conversion must respect the symmetries of Euclidean space: rotating or translating a protein does not change its internal structure. The Structure Module achieves this through Invariant Point Attention (IPA), a mechanism that works in local coordinate frames attached to each residue.
3. Why Structure Prediction Matters
3.1 Structure Determines Function
A protein’s three-dimensional shape is not decorative—it is the physical basis for everything the protein does. Enzymes accelerate reactions because their active sites cradle substrates with angstrom-level precision. Antibodies recognize pathogens because their complementarity-determining regions (CDRs) mold to antigen surfaces. Ion channels open and close because conformational changes in their transmembrane helices gate the pore. Without knowing the shape, you are guessing at mechanism.
The structure-function relationship runs deep. Two proteins with unrelated sequences but similar folds often perform similar chemistry—an observation formalized as the “structural basis of protein function.” Conversely, a single amino acid change can disrupt a fold and destroy function entirely. This is precisely what happens in many genetic diseases.
3.2 Genetic Disease: When Structure Breaks
Missense mutations—single amino acid substitutions caused by point mutations in DNA—are the most common class of pathogenic variants in the human genome. More than half of all variants classified as “pathogenic” in ClinVar are missense mutations6. Understanding why a mutation causes disease almost always requires structural reasoning.
Consider sickle cell disease. A single glutamate-to-valine substitution at position 6 of the beta-globin chain creates a hydrophobic patch on the protein surface. In the deoxygenated state, this patch binds to a complementary pocket on an adjacent hemoglobin molecule, triggering polymerization into rigid fibers that distort red blood cells. The disease mechanism is invisible without the structure.
The same logic applies across thousands of diseases. Cystic fibrosis arises from mutations in the CFTR chloride channel, many of which disrupt the fold of its nucleotide-binding domains. Familial hypercholesterolemia results from mutations in the LDL receptor that prevent proper folding or ligand binding. Predicting how a mutation reshapes a protein’s structure is the first step toward understanding—and potentially correcting—its pathological consequences.
Before AlphaFold, structural interpretation of missense variants was limited to the roughly 35% of human proteins with experimental structures. The remaining “dark proteome” was beyond reach. AlphaFold2 changed this calculus overnight: with predicted structures for nearly every human protein, variant interpretation became tractable genome-wide.
3.3 Drug Discovery: Structure-Based Design
Structure-based drug design (SBDD) is one of the oldest and most successful applications of structural biology. The logic is straightforward: if you know the three-dimensional shape of a drug target—especially its binding pocket—you can computationally screen or design molecules that fit.
HIV protease inhibitors are the textbook example. In the early 1990s, crystal structures of HIV-1 protease revealed the geometry of its active-site cleft. Medicinal chemists designed inhibitors (saquinavir, ritonavir, indinavir) that precisely complemented the pocket’s shape and electrostatics, transforming AIDS from a death sentence into a manageable chronic condition7.
SBDD historically required experimental structures—months of protein expression, purification, crystallization, and data collection. AlphaFold2 predictions now provide starting models for targets that have resisted crystallization. Several pharmaceutical companies have reported using AlphaFold structures for:
- Binding pocket identification. Finding druggable cavities on targets with no experimental structure, particularly membrane proteins and intrinsically flexible targets.
- Virtual screening. Docking millions of compounds against predicted structures to identify candidate binders, then validating top hits experimentally.
- Lead optimization. Understanding how a lead compound interacts with its target at the atomic level, guiding chemical modifications that improve potency and selectivity.
- Allosteric site discovery. Identifying secondary binding sites distant from the active site, which can modulate protein function through conformational changes.
The accuracy threshold matters here. Drug design requires structures accurate to 1–2 angstroms in the binding pocket. AlphaFold2 predictions with pLDDT scores above 90 typically meet this standard for ordered regions, though predictions of flexible loops around binding sites remain less reliable.
3.4 Antibody Engineering
Antibodies bind their targets through six hypervariable loops (CDRs) arranged on a relatively rigid framework. Predicting the conformations of these loops—especially CDR-H3, the most structurally diverse—has been a major challenge in computational immunology.
AlphaFold2 and its successors have substantially improved CDR loop prediction. This matters for antibody engineering because:
- Humanization. Grafting CDR loops from animal antibodies onto human frameworks requires knowing how the loops will sit on the new scaffold.
- Affinity maturation. Predicting how mutations in the CDR loops alter the binding interface guides rational library design.
- Epitope mapping. Predicting the structure of an antibody-antigen complex reveals which residues on the antigen are contacted, informing vaccine design and diagnostic development.
AlphaFold-Multimer (discussed in Section 4) can predict antibody-antigen complexes directly, though accuracy for these interactions remains lower than for other protein-protein complexes due to the extreme sequence diversity of CDR loops.
3.5 Functional Annotation at Scale
The most transformative application of structure prediction may be functional annotation of the “dark proteome.” Before AlphaFold2, approximately 170,000 protein structures were available in the Protein Data Bank—representing less than 0.1% of known protein sequences. For most proteins, the only available information was the amino acid sequence.
Structure enables function prediction through several routes:
- Fold-level homology. Two proteins with similar folds are likely related by distant evolution, even if their sequences have diverged beyond recognition. Structure comparison tools like Foldseek and DALI can identify these relationships in seconds.
- Active site identification. Conserved spatial arrangements of catalytic residues (catalytic triads, metal-binding sites, cofactor-binding pockets) reveal enzymatic function.
- Interaction interfaces. Predicted surface properties—hydrophobic patches, charged grooves, beta-sheet edges—suggest binding partners and interaction modes.
- Domain decomposition. Multi-domain proteins can be decomposed into structural modules, each with a recognizable function.
Metagenomic sequencing has uncovered millions of protein sequences from uncultured organisms. These sequences have no experimental characterization whatsoever. Structure prediction allows computational functional annotation at a scale that was previously impossible, opening access to the biochemical diversity of the environmental microbiome.
3.6 Understanding Resistance Mutations
In infectious disease and oncology, drug resistance arises when mutations in the target protein reduce drug binding without abolishing the protein’s native function. Structural analysis reveals how resistance mutations work: do they directly block the drug’s binding site, alter the shape of a pocket, or shift the protein’s conformational equilibrium?
Kinase inhibitor resistance in cancer provides a vivid example. The T790M mutation in EGFR (epidermal growth factor receptor) confers resistance to first-generation tyrosine kinase inhibitors like erlotinib by introducing a bulky methionine side chain that sterically clashes with the drug. Understanding this structural mechanism led to the development of third-generation inhibitors (osimertinib) designed to accommodate the mutation8.
With structure prediction, resistance mutation analysis no longer depends on solving a new crystal structure for each mutant. Predicted structures of mutant proteins can be generated in minutes, enabling rapid structural interpretation of clinically observed resistance.
4. How People Use Structure Prediction Today
4.1 The AlphaFold Protein Structure Database
In July 2021, DeepMind and the European Bioinformatics Institute (EMBL-EBI) released the AlphaFold Protein Structure Database (AFDB), initially covering 365,000 protein structures across 21 model organisms9. By the end of 2022, the database expanded to over 200 million predicted structures, covering essentially every protein in UniProt—the comprehensive database of known protein sequences.
This represents a qualitative shift. Before AFDB, obtaining a protein structure required either solving it experimentally (months to years of work, with a significant failure rate) or running AlphaFold2 yourself (requiring substantial GPU resources and bioinformatics expertise). Now, for most proteins, the structure is already predicted and downloadable in seconds.
The database has been accessed over 10 million times and cited in thousands of studies. Typical use cases include:
- Browsing a predicted structure for a protein of interest to generate hypotheses about its function, binding sites, or interaction interfaces.
- Downloading coordinates for use as starting models in molecular replacement for X-ray crystallography or cryo-EM.
- Feeding predicted structures into downstream computational pipelines (docking, molecular dynamics, functional annotation).
4.2 ColabFold: Fast, Accessible Predictions
ColabFold (Mirdita et al., 2022) wraps AlphaFold2 in a user-friendly Google Colab notebook that runs in a web browser10. Its key innovation is replacing the slow JackHMMER-based MSA search with MMseqs2, a much faster sequence search tool. This reduces the total prediction time from hours to minutes for most proteins.
ColabFold democratized structure prediction further by eliminating hardware requirements. A researcher with nothing more than a web browser and a protein sequence can obtain a predicted structure in under 30 minutes, including MSA generation and model inference. It also supports batch predictions, custom MSAs, and AlphaFold-Multimer for protein complexes.
4.3 ESMFold: Single-Sequence Prediction
ESMFold (Lin et al., 2023) takes a fundamentally different approach. Instead of building an MSA from sequence databases, ESMFold uses the embeddings from ESM-2, a protein language model trained on millions of individual sequences (covered in Lecture 5). The language model’s internal representations encode enough evolutionary information—learned implicitly from patterns across the training set—to predict structure from a single sequence11.
This matters in two scenarios:
- Speed. ESMFold predictions are roughly 60 times faster than AlphaFold2 because they skip the MSA construction step entirely. For proteome-scale screening, this speed difference is decisive.
- Orphan proteins. Some proteins have few detectable homologs in sequence databases—novel viral proteins, de novo designed sequences, or proteins from poorly sampled branches of the tree of life. ESMFold can still make predictions for these, using the implicit evolutionary knowledge in its language model.
The trade-off is accuracy. For well-studied protein families with deep MSAs, AlphaFold2 remains more accurate. For proteins with shallow or no MSA, ESMFold’s language model representations provide a viable alternative.
4.4 AlphaFold3: Beyond Single Chains
AlphaFold3 (Abramson et al., 2024) extends the AlphaFold paradigm from single protein chains to arbitrary biomolecular complexes. It can predict the structures of:
- Protein-protein complexes (already possible with AlphaFold-Multimer, but improved)
- Protein-DNA and protein-RNA complexes
- Protein-ligand complexes (small molecules, ions, cofactors)
- Post-translational modifications (phosphorylation, glycosylation)
The architectural changes from AlphaFold2 to AlphaFold3 are substantial. AlphaFold3 replaces the Evoformer with a Pairformer (which processes only the pair representation, not a full MSA stack) and replaces the deterministic Structure Module with a diffusion-based structure generation module. These changes improve the model’s ability to handle multiple molecular types and generate diverse conformational samples.
AlphaFold3’s predictions for protein-ligand interactions are particularly significant for drug discovery, though accuracy for small-molecule binding poses remains below what dedicated docking tools achieve for many systems.
4.5 Practical Workflows: Choosing the Right Tool
Different prediction tools suit different scenarios. The choice depends on accuracy requirements, speed constraints, and the nature of the target.
| Scenario | Recommended tool | Rationale |
|---|---|---|
| Single protein, high accuracy needed | AlphaFold2 (ColabFold) | Best accuracy for proteins with deep MSAs |
| Proteome-scale survey | ESMFold | 60x faster, no MSA construction needed |
| Protein-protein complex | AlphaFold-Multimer | Specialized for inter-chain contacts |
| Protein-ligand complex | AlphaFold3 | Handles small molecules natively |
| Designed (non-natural) protein | ESMFold or AlphaFold2 | ESMFold if no homologs; AF2 if related sequences exist |
| Membrane protein | AlphaFold2 | Generally reliable for transmembrane regions with pLDDT > 70 |
| Experimental model building | AlphaFold2 → MR/cryo-EM | Use predicted structure as starting model for experimental phasing |
4.6 Interpreting pLDDT Scores
Every AlphaFold prediction comes with per-residue confidence scores. The predicted Local Distance Difference Test (pLDDT) measures how confident the model is about each residue’s local environment12.
| pLDDT range | Interpretation | Typical use |
|---|---|---|
| > 90 | Very high confidence | Backbone and side-chain positions reliable; suitable for structure-based drug design |
| 70–90 | Confident | Backbone fold is correct; side-chain orientations may be approximate |
| 50–70 | Low confidence | General topology may be correct, but details are unreliable |
| < 50 | Very low confidence | Likely intrinsically disordered or flexible; no single structure is meaningful |
Critically, low pLDDT does not always mean the prediction is wrong—it can also indicate genuine biological disorder. Intrinsically disordered regions (IDRs), which lack a stable 3D structure in vivo, consistently receive low pLDDT scores. This has turned AlphaFold2 into an unexpectedly powerful disorder predictor.
The predicted aligned error (PAE) matrix provides complementary information about the relative positions of residue pairs. High PAE between two domains suggests that AlphaFold is confident about each domain’s internal structure but uncertain about their relative orientation—a common situation for multi-domain proteins connected by flexible linkers.
4.7 AlphaFold-Multimer for Protein Complexes
Protein function frequently depends on interactions between multiple chains. Hemoglobin is a tetramer; the ribosome assembles from dozens of proteins and RNA molecules; signaling cascades depend on transient protein-protein interactions.
AlphaFold-Multimer extends the AlphaFold2 architecture to predict the structures of protein complexes. The key modification is constructing paired MSAs that align homologous sequences across chains from the same organism. If chain A and chain B always co-occur in the same species, their co-evolutionary patterns reveal inter-chain contacts—the same logic that identifies intra-chain contacts from standard MSAs.
Practical considerations for multimer predictions:
- Accuracy is generally highest for stable, obligate complexes (subunits that always function together).
- Transient interactions (signaling complexes, enzyme-substrate) are harder because the evolutionary signal is weaker.
- Confidence is assessed from the inter-chain PAE: low values indicate high confidence in the predicted interface geometry.
- Antibody-antigen complexes remain challenging because the extreme diversity of CDR loops limits co-evolutionary signal.
4.8 Integration with Other Computational Methods
Predicted structures are rarely endpoints. They feed into downstream computational workflows:
Molecular docking. Predicted structures serve as receptors for virtual screening of drug candidates. Tools like AutoDock Vina, Glide, and DiffDock accept AlphaFold coordinates as input. Regions with high pLDDT are treated as reliable; low-confidence loops near binding sites may need to be modeled with conformational sampling.
Molecular dynamics (MD) simulations. AlphaFold predictions provide starting configurations for MD runs that explore conformational dynamics, binding thermodynamics, and allosteric pathways. The static AlphaFold prediction represents one snapshot; MD generates the movie.
Protein design. Structure prediction is a key component of the design-build-test cycle. Tools like ProteinMPNN (Lecture 10) use predicted or designed backbone structures as input for sequence optimization. RFDiffusion (Lecture 9) generates novel backbones conditioned on functional constraints, and AlphaFold validates whether the designed sequences are predicted to fold as intended.
Functional annotation. Structure comparison tools like Foldseek search the AFDB for structurally similar proteins, enabling functional transfer from characterized proteins to uncharacterized ones at a scale not possible with sequence-based methods alone.
4.9 Limitations
Structure prediction has not solved all of structural biology. Several important limitations remain:
Conformational dynamics. AlphaFold2 predicts a single static structure for each protein. Many proteins undergo functionally important conformational changes—allosteric transitions, domain motions, ligand-induced fit. The predicted structure typically represents the most stable conformation in the training data, which may not be the functionally relevant state.
Intrinsically disordered regions. Approximately 30% of eukaryotic proteins contain intrinsically disordered regions that do not adopt a single stable fold. AlphaFold2 assigns low pLDDT to these regions (correctly indicating uncertainty), but this also means it provides no useful structural information for a substantial fraction of the proteome.
Novel folds with few homologs. AlphaFold2’s accuracy depends heavily on the depth and diversity of the input MSA. For proteins with very few homologs—orphan proteins from poorly sampled organisms, de novo designed sequences, or recently evolved viral proteins—prediction accuracy can drop substantially.
Protein-ligand interactions. While AlphaFold3 can predict protein-ligand complexes, the accuracy of binding pose prediction and affinity ranking remains below what is needed for reliable lead optimization without experimental validation.
Mutations and variants. AlphaFold2 was trained on wild-type sequences from the PDB. While it can accept mutant sequences as input, it was not specifically trained to model the structural effects of point mutations, and its predictions for mutant proteins are not always more informative than those for the wild type.
5. Design Principles and Lessons
AlphaFold2’s success was not accidental. Every component reflects principled thinking about what information matters for protein structure and how neural networks can extract it.
Principle 1: Use Evolutionary Information
Proteins are not designed from scratch; they evolve from ancestors. Related sequences are experiments run by natural selection, each revealing something about the constraints on structure. AlphaFold2 makes MSA processing central, with specialized attention mechanisms that extract co-evolutionary signals.
Principle 2: Model Pairwise Relationships Explicitly
Protein structure is fundamentally about which residues are near which other residues. The pair representation makes these relationships a first-class data structure, and triangular operations enforce that pairwise predictions are geometrically self-consistent.
Principle 3: Respect Symmetry
Three-dimensional space has rotational and translational symmetry. Rather than hoping the network discovers this from data, AlphaFold2 builds invariance into the architecture through local frames and IPA. This is an instance of a broader principle: inductive biases that match the problem domain reduce the burden on learning.
Principle 4: Iterate and Refine
The Evoformer runs 48 blocks. The Structure Module runs 8 iterations. The full pipeline is recycled 3 times. Each pass improves on the last, allowing local decisions to propagate globally and global context to inform local geometry.
Principle 5: Learn What You Do Not Know
The pLDDT confidence head is not an afterthought. AlphaFold2 explicitly models its own uncertainty, distinguishing confident predictions from guesses. This has proven valuable for downstream users: a prediction with pLDDT > 90 can be trusted for atomic-level analysis, while regions below 50 are likely disordered or poorly predicted.
| Principle | Implementation |
|---|---|
| Use evolutionary information | MSA processing in Evoformer |
| Model pairwise relationships | Pair representation + triangular updates |
| Respect symmetry (SE(3)) | IPA, frame-based representation |
| Iterate and refine | 8 IPA iterations, 3 recycling passes |
| Multi-task learning | FAPE + distogram + pLDDT + masked MSA |
| Calibrated confidence | pLDDT head trained against actual local accuracy |
Key Takeaways
-
Structure prediction was a 50-year grand challenge. From Anfinsen’s hypothesis (1972) through Levinthal’s paradox and 25 years of CASP competitions, predicting a protein’s 3D structure from its amino acid sequence resisted all approaches—until AlphaFold2 crossed the experimental accuracy threshold at CASP14 in 2020.
-
AlphaFold2 crossed the experimental accuracy threshold. A median GDT-TS above 90 at CASP14 demonstrated that deep learning could match the accuracy of X-ray crystallography for most targets, using evolutionary information and learned geometric reasoning.
-
Structure determines function. Knowing a protein’s shape enables understanding genetic diseases (missense mutations disrupting folds), structure-based drug design (targeting binding pockets), functional annotation of unknown proteins, and interpretation of resistance mutations.
-
200 million predicted structures are democratizing structural biology. The AlphaFold Protein Structure Database covers essentially all known proteins. ColabFold makes predictions accessible through a browser. ESMFold provides fast single-sequence predictions from protein language model embeddings.
-
pLDDT scores indicate prediction reliability. Scores above 90 are reliable at atomic resolution; scores below 50 typically indicate intrinsic disorder. The PAE matrix reveals inter-domain confidence. These scores guide which regions to trust for downstream applications.
-
Important limitations remain. Conformational dynamics, intrinsically disordered regions, proteins with few homologs, and accurate protein-ligand binding pose prediction all represent open challenges.
-
Anfinsen’s thermodynamic hypothesis is sometimes called the “thermodynamic control” model of folding, as opposed to kinetic control, where folding intermediates might trap the protein in a non-native state. ↩
-
Denaturation is the loss of a protein’s three-dimensional structure. Heat, extreme pH, or chemical agents (like urea) disrupt the non-covalent interactions — hydrogen bonds, hydrophobic packing, salt bridges — that hold the fold together, leaving the chain as a floppy, unstructured polymer. ↩
-
The “folding funnel” picture, introduced by Wolynes, Onuchic, and Thirumalai in the 1990s, describes the free-energy landscape as a rugged funnel in which the native state sits at the bottom. ↩
-
The GDT-TS (Global Distance Test - Total Score) metric measures the fraction of residues whose C\(_\alpha\) atoms fall within various distance cutoffs of the true structure after optimal superposition. A score above 90 is generally considered comparable to experimental accuracy for medium-resolution crystal structures. ↩
-
A multiple sequence alignment (MSA) is a matrix where each row is a related protein sequence and each column aligns evolutionarily corresponding positions. MSAs are built by search tools like JackHMMER or HHBlits, which scan large sequence databases to find homologs of the query protein. A typical MSA for AlphaFold2 may contain thousands of sequences. ↩
-
ClinVar is a public database maintained by NCBI that aggregates information about the relationship between human genetic variants and phenotypes, including disease associations and clinical significance classifications. ↩
-
Structure-based design of HIV protease inhibitors is one of the landmark successes of rational drug design. The initial crystal structures were solved by groups at Merck, Abbott, and Roche in the late 1980s and early 1990s, and the first protease inhibitor (saquinavir) was approved by the FDA in 1995. ↩
-
The T790M “gatekeeper” mutation in EGFR is the most common mechanism of acquired resistance to first- and second-generation EGFR inhibitors in non-small cell lung cancer. Structural analysis of the mutant kinase domain guided the design of covalent inhibitors that bind an alternative cysteine residue. ↩
-
The AlphaFold Protein Structure Database is freely available at alphafold.ebi.ac.uk. Each entry includes the predicted 3D coordinates, per-residue pLDDT confidence scores, and predicted aligned error (PAE) matrices. ↩
-
ColabFold is available at github.com/sokrypton/ColabFold. It supports both AlphaFold2 and ESMFold backends and can run on free Google Colab GPUs. ↩
-
ESMFold achieves competitive accuracy with AlphaFold2 for many proteins, though accuracy drops for proteins with few homologs in ESM-2’s training data. Its primary advantage is speed: predictions take seconds rather than minutes. ↩
-
pLDDT is a per-residue score between 0 and 100. It is trained to predict the actual lDDT-C\(_\alpha\) score, which measures the fraction of distances between a residue and its neighbors (within 15 angstroms) that are predicted within specified thresholds. It is not the same as B-factor, though the two correlate for ordered regions. ↩