Note: For background on gas adsorption, GCMC, and classical DFT, see our earlier adsorption tutorial.

Grand canonical Monte Carlo (GCMC) is the gold-standard simulation for gas adsorption, used to design materials from methane storage to CO₂ direct air capture. It samples gas molecules in the material one at a time, averaging them into the equilibrium adsorbate density field — accurate, but far too slow to screen candidates at scale. In our recent work, we introduce MADField, which evaluates adsorption about 250,000× faster than GCMC with no loss in accuracy. The speedup comes from a change of paradigm: instead of simulating particles, MADField predicts the same density field with a neural network trained at two fidelities — pre-trained on cheap, abundant classical DFT (cDFT) and fine-tuned on a little high-fidelity GCMC — then integrates it for uptake. On all 270,583 ARC-MOF structures, MADField recovers 95% of the rare high-capacity materials within the top 1.7% of its ranking, 56× more precisely than the best previous ML baseline.
The full method and experiments are in our paper, MADField (arXiv:2606.21284).
Material “adsorbent” discovery
Adsorption is the process by which gas molecules accumulate inside the pores of a porous solid. The solid that holds the gas is the adsorbent; the gas it holds is the adsorbate. Methane inside a porous crystal, or carbon dioxide pulled from flue gas, are both adsorption.
Finding good adsorbents is one of the large axes of materials discovery. Porous materials like zeolites, metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and amorphous carbons are all actively explored. The 2025 Nobel Prize in Chemistry, awarded to Robson, Kitagawa, and Yaghi for the design and synthesis of MOFs (Nobel Prize in Chemistry, 2025), reflects how central this class of materials has become.
These porous materials span an enormous space — hundreds of thousands synthesized, and millions more proposed computationally (Wilmer et al., 2012) — so the chance that an excellent adsorbent exists somewhere in it is high. The difficulty is finding it: a needle-in-a-haystack search over a space too large. This is the central problem of adsorbent discovery.

Bottleneck: evaluating candidates in scale
Evaluating a candidate means computing its gas uptake $N$: the amount of gas it adsorbs at a given temperature and pressure. Uptake is the single number used to rank materials in high-throughput screening, since most applications come down to storing or capturing as much gas as possible at a target condition.
The catch is that we can’t read uptake off directly. To know how much gas a material holds, we first have to know where it all goes — the full 3D pattern of gas in the pores, its density field $\rho(\mathbf{r})$. Uptake $N$ is just the integral of that field over the unit cell,
\[N(P, T) = \int_{\mathcal{V}} \rho_{\mathrm{eq}}(\mathbf{r})\, d\mathbf{r},\]so the real work is getting $\rho(\mathbf{r})$ — and there are two standard ways, both slow:
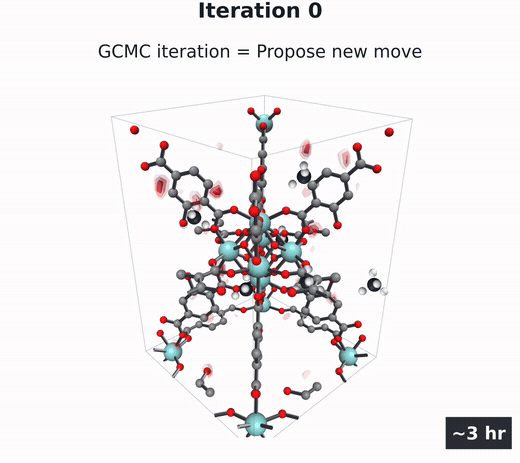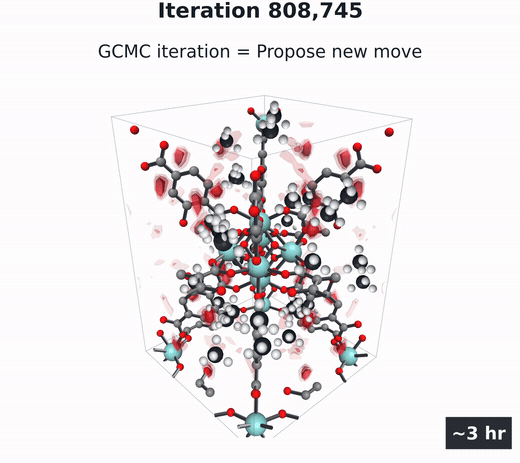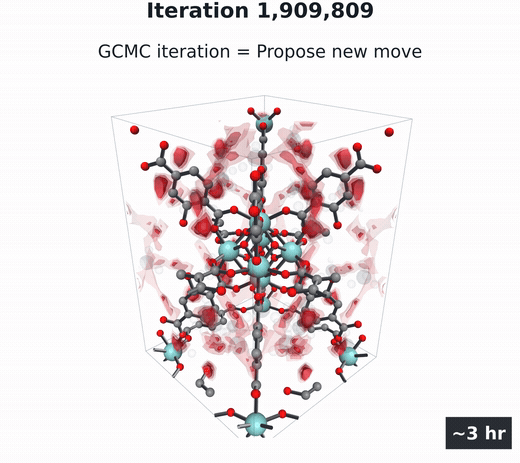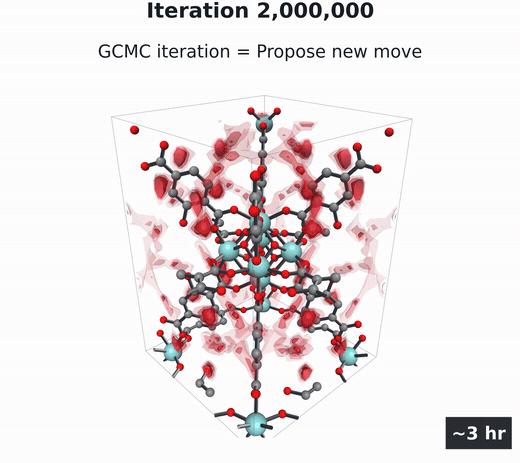
GCMC
Molecules build up and move, then coarse-grain into the density field. Millions of steps.
cDFT
Propose a new density to minimize the free-energy functional. Tens–hundreds of iterations.
- Grand canonical Monte Carlo (GCMC) (Adams, 1975; Dubbeldam et al., 2016) — the particle-based gold standard. Randomly insert, delete, and move gas molecules in the pores over millions of steps, then average the snapshots into $\rho(\mathbf{r})$. Accurate, but several hours per material per condition.
- Classical density functional theory (cDFT) — skip the particles and solve for $\rho(\mathbf{r})$ directly, minimizing a free-energy functional (Evans, 1979) by fixed-point iteration. Faster, but rests on an approximate functional and still iterates for every case.
A practical screening covers hundreds of thousands of materials, and even cDFT — the faster of the two — costs roughly 5 GPU-minutes per material at a fixed temperature and pressure, so screening 100,000 candidates runs to about 350 GPU-days. GCMC would take far longer even with multicore CPU. And those hundreds of thousands are only the experimental structures; the space of hypothetical frameworks proposed in silico runs from millions to trillions, where brute-force simulation is hopeless from the start.
MADField predicts $\rho(\mathbf{r})$ directly
MADField is a neural network that predicts the 3D equilibrium gas density field in porous materials. Given a material, an adsorbate, and a thermodynamic condition, it outputs the full density field $\rho(\mathbf{r})$ in sub-second — and gas uptake $N$ is computed by integration.

Result 1: benchmark on a practical MOF screening pipeline
For methane storage the quantity that matters is working capacity (WC): the CH₄ a material actually delivers between a full and an empty tank — its uptake swing between the storage and depletion pressures,
\[\mathrm{WC} = N(P_{\mathrm{ads}}, T) - N(P_{\mathrm{des}}, T),\]with $P_{\mathrm{ads}} = 65~\mathrm{bar}$, $P_{\mathrm{des}} = 5.8~\mathrm{bar}$, and $T = 298~\mathrm{K}$, so each candidate needs two uptake evaluations. We call a framework a high-capacity target when its $\mathrm{WC} \ge 200~\mathrm{cm^3/cm^3}$.
Across the full ARC-MOF database (Burner et al., 2023) of 270,583 frameworks, only 167 clear that bar — 0.06% of the database, a true needle in a haystack. Ranking all of them by predicted WC, MADField recovers 95% of the targets within the top 1.7% of candidates, at 56× higher average precision1 than the best learned baseline,2 and five orders of magnitude less cost than GCMC.

Result 2: uptake accuracy on unseen materials
MADField generalizes to materials it never saw during training. On in-distribution MOFs every model does reasonably well — MADField tracks the reference isotherm (uptake versus pressure at a fixed temperature) most closely, but strong baselines such as Uni-MOF (Wang et al., 2024) stay near it too. The picture splits on unseen materials: for disordered solids like amorphous carbons (Thyagarajan & Sholl, 2020) — along with polymers of intrinsic microporosity (PIMs), hyper-cross-linked polymers, and kerogens3 — MADField still follows the reference closely, while the baselines break down badly.

Result 3: density accuracy
MADField predicts the density field itself accurately, not just its integral. The mean Tanimoto similarity to the reference density is 0.996 (cDFT) and 0.965 (GCMC) across MOFs — versus 0.943 and 0.891 for the strongest density baseline, DeepAPD (Burner et al., 2026) — and on unseen amorphous carbons the margin widens to 0.885 against 0.492.

Result 4: acceleration of cDFT
Beyond replacing simulation, MADField can also speed it up. Because it predicts an unnormalized density in physical units, that prediction is a ready-made starting guess for the cDFT fixed-point solver: warm-starting from it cuts solver iterations by 2.0× and recovers convergence in 42% of cases that fail from the standard initialization.
Two ideas behind MADField’s success
Two ideas drive these results. The first is the prediction target: MADField outputs the full density field $\rho(\mathbf{r})$ rather than the scalar uptake $N$ — a single object that yields uptake, binding sites, and isotherms together, and the source of most of the accuracy. The second is how it is trained: a broad, cheap cDFT prior corrected by a little high-fidelity GCMC, which is what lets it generalize to materials it never saw. We take each in turn.
$\rho(\mathbf{r})$ as a unified target, not the scalar $N$
Most ML models for adsorption predict scalar uptake \(N\) directly. We predict the full 3D equilibrium gas density field \(\rho(\mathbf{r})\) instead, and recover uptake by integrating it:
\[N = \int_{\mathcal{V}} \rho(\mathbf{r})\,d\mathbf{r}\]Integrating the field recovers uptake, but it carries more than its integral: where \(\rho(\mathbf{r})\) concentrates marks the binding sites, and sweeping pressure traces the full isotherm. All three fall out of one prediction.
A version of MADField that shares the exact same backbone but regresses \(N\) directly is 7.2× worse. The accuracy gain comes from the target, not the network.
Density-field prediction is not new (Sun & Siepmann, 2024; Burner et al., 2026), but prior work has a critical gap: existing models predict a normalized density that integrates to one by construction. Integrating it gives 1, not uptake. Some models require the uptake \(N\) itself as an input to work around this. Neither can independently predict how much gas a material holds.
MADField predicts an unnormalized density in physical units (molecules per ų). Integrating it gives the real uptake — no normalization trick, no uptake fed in.
Multi-fidelity: cDFT teaches, GCMC corrects
Predicting a density field rather than a scalar makes the data problem harder: we need density-field labels, not just uptake numbers. GCMC-derived density fields are the accurate target, but expensive to generate at scale. cDFT fields are cheap and abundant, but inherit the approximation error of the cDFT functional.
We use both. MADField is first pre-trained on 280,000 cDFT calculations spanning 4,000 MOFs and nine adsorbates — we call this cDFT-only model MADField-cDFT — then fine-tuned on a GCMC dataset 14.7× smaller to give MADField-GCMC. cDFT teaches a broad prior — how geometry, adsorbate identity, and thermodynamic conditions shape the density field. GCMC corrects the remaining fidelity gap.

How good is the cheap, approximate cDFT data on its own? Good enough that MADField-cDFT — trained without a single GCMC label — already outranks every learned baseline on the screening above, and on per-MOF working-capacity accuracy it places second only to MADField-GCMC, ahead of Uni-MOF and the rest. Starting from that strong cDFT foundation, the small GCMC set then delivers a decisive gain: fine-tuning raises screening average precision 8× (MADField-cDFT → MADField-GCMC, 0.068 → 0.557). cDFT supplies the breadth, GCMC the fidelity — the full result needs both.

This is what drives generalization. We also tried training on GCMC alone, without the cDFT prior, and performance dropped sharply — most of all on out-of-distribution materials. The cDFT prior is what carries MADField to material classes it never saw during fine-tuning.
Conclusion
MADField reframes adsorption prediction as estimating the 3D equilibrium density field, then recovers gas uptake by integrating it. That one target unifies what a screening pipeline needs: ranking candidates by working capacity, accurate uptake on seen and unseen materials, faithful density fields, and a warm start that accelerates the cDFT solver itself. Multi-fidelity training is what makes it work — a broad, cheap cDFT prior corrected by a little high-fidelity GCMC — and what carries the model from MOFs to disordered solids it never trained on.
MADField generalizes far beyond the MOFs it was trained on — to amorphous carbons and other disordered porous materials, including frameworks with thousands of atoms and unit cells far larger than anything it saw in training, where the strongest baselines fall apart but MADField stays accurate. Its predicted density also gives cDFT a strong initial guess, cutting the iterations to convergence by about half and recovering many cases that otherwise fail to converge at all.
Real adsorbent discovery is a needle-in-a-haystack retrieval task. Our database-scale screening benchmark measures whether a model surfaces the rare high-capacity materials, not merely whether it reproduces the overall trend. Average uptake error is too weak a metric for a deployable screening pipeline. Predictions must rank the best materials at the top, and MADField is the first model in this benchmark to clear that bar by a wide margin.
References
- Y. Kim, S. Kim, S. Ahn, H. Kim. (2026). MADField: Multi-fidelity Amortized Density Field for Adsorption in Nanoporous Materials. arXiv:2606.21284.
- Burner, J., et al. (2023). ARC-MOF: A Diverse Database of Metal-Organic Frameworks with DFT-Derived Partial Charges and Descriptors for Enhanced Machine Learning Predictions. Chemistry of Materials. ↩
- Burner, J., Marchand, O., Cicciarella, R., Gibaldi, M. & Woo, T. K. (2026). Rapid prediction of single-site adsorbate probability distributions in metal–organic frameworks using graph neural networks (DeepAPD). Digital Discovery. ↩
- Sun, Y. & Siepmann, J. I. (2024). Understanding and predicting the spatially resolved adsorption properties of nanoporous materials (SorbIIT). Journal of Chemical Theory and Computation, 20. ↩
- Wang, J., et al. (2024). A comprehensive transformer-based approach for high-accuracy gas adsorption predictions in metal–organic frameworks (Uni-MOF). Nature Communications, 15. ↩
- Kang, Y., Park, H., Smit, B. & Kim, J. (2023). A multi-modal pre-training transformer for universal transfer learning in metal–organic frameworks (MOFTransformer). Nature Machine Intelligence, 5. ↩
- Cui, J., et al. (2023). Direct prediction of gas adsorption via spatial atom interaction learning (DeepSorption). Nature Communications, 14. ↩
- Sarikas, A. P., Gkagkas, K. & Froudakis, G. E. (2024). Gas adsorption meets deep learning: voxelizing the potential energy surface of metal–organic frameworks (RetNet). Scientific Reports, 14. ↩
- Lin, E., Zhong, Y., Chen, G. & Deng, S. (2025). Unified physio-thermodynamic descriptors via learned CO₂ adsorption properties in metal–organic frameworks (IsothermNet). npj Computational Materials, 11. ↩
- Evans, R. (1979). The nature of the liquid-vapour interface and other topics in the statistical mechanics of non-uniform, classical fluids. Advances in Physics, 28(2):143–200. ↩
- Thyagarajan, R. & Sholl, D. S. (2020). A Database of Porous Rigid Amorphous Materials. Chemistry of Materials. ↩
- The Royal Swedish Academy of Sciences. (2025). The Nobel Prize in Chemistry 2025 — Susumu Kitagawa, Richard Robson, Omar M. Yaghi. NobelPrize.org. ↩
- Wilmer, C. E., et al. (2012). Large-scale screening of hypothetical metal–organic frameworks. Nature Chemistry, 4. ↩
- Adams, D. J. (1975). Grand canonical ensemble Monte Carlo for a Lennard-Jones fluid. Molecular Physics, 29. ↩
- Dubbeldam, D., Calero, S., Ellis, D. E. & Snurr, R. Q. (2016). RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Molecular Simulation, 42. ↩
- Rosen, J., et al. (2021). Machine learning the quantum-chemical properties of metal–organic frameworks for accelerated materials discovery (QMOF). Matter.
-
Average precision summarizes how well a ranking surfaces rare positives near the top. It is the area under the precision–recall curve, so a higher value means the high-capacity targets appear earlier in the screened list. ↩
-
Learned uptake baselines: Uni-MOF (Wang et al., 2024), MOFTransformer (Kang et al., 2023), DeepSorption (Cui et al., 2023), RetNet (Sarikas et al., 2024), and IsothermNet (Lin et al., 2025); density-field baselines: DeepAPD (Burner et al., 2026) and SorbIIT (Sun & Siepmann, 2024). ↩
-
Kerogen is the disordered, insoluble organic matter dispersed in sedimentary rock — the precursor to oil and gas, and a natural microporous adsorbent whose pore structure differs sharply from a crystalline MOF. ↩